
您好,欢迎来到虫洞时空!
请登录
[买家]
[卖家]
|
免费注册
买家注册
卖家注册
| 品牌 | 宝杰罗 | |
| 基础属性 | 生产企业 | 北京宝杰罗生物公司 |
| cas号 | ||
| 规格 | 个样品 | |
| 储存条件 | ||
| 纯度 | ||
| 密度 |
菌群组成谱全长测序研究
基于PacBio三代测序平台,能够轻而易举地读取群落微生态系统中所有微生物的rRNA基因/ITS全长序列,不再受制于短序列的局限性,并充分保障全长序列的测序精确性,从而在种甚至菌株等精细水平更全面更深入地解析菌群多样性和组成谱。分析出的微生物菌群多样性比二代短序列的高2-3倍。
产品特色
l 深入获取微生物群落的精细组成信息;
l 预测微生物群落的潜在代谢功能,并指导后续宏基因组测序研究;
l 通过“全微生物组关联研究(Microbiome-Wide Association Studies,MWAS)”,精准鉴定与表型/组间差异相关的关键物种。
应用领域
l 微生物组与环境互作关系的研究;
l 微生物组与宿主共生关系的研究;
l 微生物组在临床医学和精准医疗领域的应用;
l 微生物组在发酵工艺、食品加工/检测等行业的应用。
测序优势
l 自动化、专业、完善的DNA提取扩增体系和样本前处理流程;
l 拥有PacBio RSII和Sequel两种单分子实时测序平台,提供一站式测序服务;
l Greengenes/Silva/HOMD/RDP/Unite/MaarjAM等多个物种注释数据库,最优化物种的分类鉴定;
l 专家教授全方位支持,结果直接用于发表,可按需个性化定制;
l 项目经验丰富全面,涵盖水样、活性污泥、土壤、排泄物、体液、组织等各类样本。
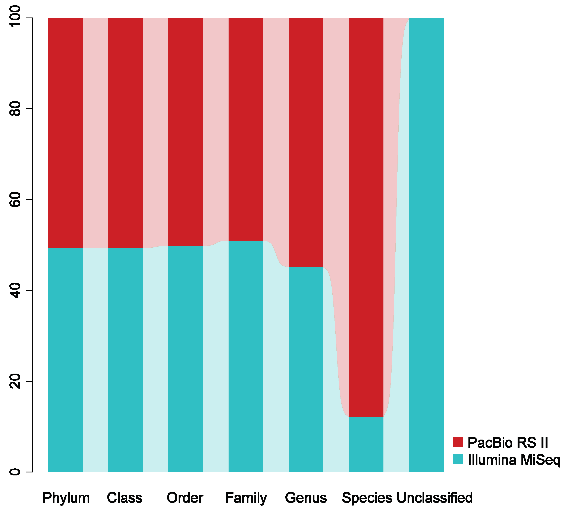

PacBio和Illumina平台对菌群16S rRNA基因测序结果的比较,三代测序的结果能显著提升种水平的物种检测精确性,并大幅改善物种分类信息的不确定性。
送样要求
|
样本 |
样本要求 |
|
土壤类 |
500 mg |
|
排泄物类 |
固体500 mg,液体200 ml |
|
水样类 |
500 ml液体过滤的滤膜 |
|
组织 |
200 mg |
|
微生物基因组总DNA |
体积> 20 ml;浓度> 20 ng/ml |
送样原则:新鲜、足量,请提供三次实验所需用量。
菌群组成谱全长测序分析流程
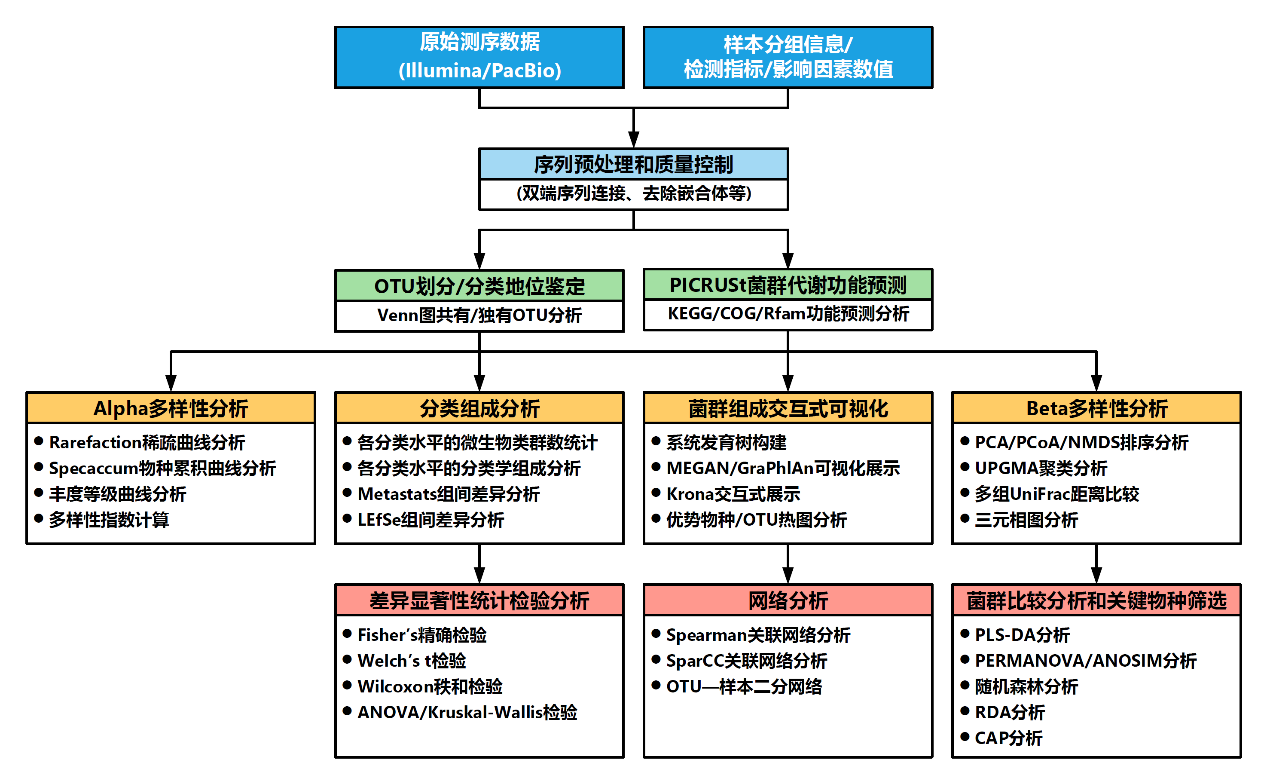





菌群多样性组成谱全长分析 ,PacBio第三代全长测序常用引物:
细菌16S rDNA:
(1) 对16S rDNA的全长进行PCR扩增
(2) 以扩增产物为模板进行PacBio测序文库的制备
(3) PacBio系统测序16S rDNA的全长序列
(4) 引物信息:27F AGAGTTTGATCMTGGCTCAG
1492R ACCTTGTTACGACTT
古菌16S rDNA:
(1) 对16S rDNA的全长进行PCR扩增
(2) 以扩增产物为模板进行PacBio测序文库的制备
(3) PacBio系统测序16S rDNA的全长序列
(4) 引物信息:21F TTCCGGTTGATCCYGCCGGA
1492R ACCTTGTTACGACTT
真菌ITS全长:
(1) 对真菌ITS的全长进行PCR扩增
(2) 以扩增产物为模板进行PacBio测序文库的制备
(3) PacBio系统测序真菌ITS的全长序列
(4) 引物信息:ITS1F CTTGGTCATTTAGAGGAAGTAA
LR3 CCGTGTTTCAAGACGGG
特色分析内容举例
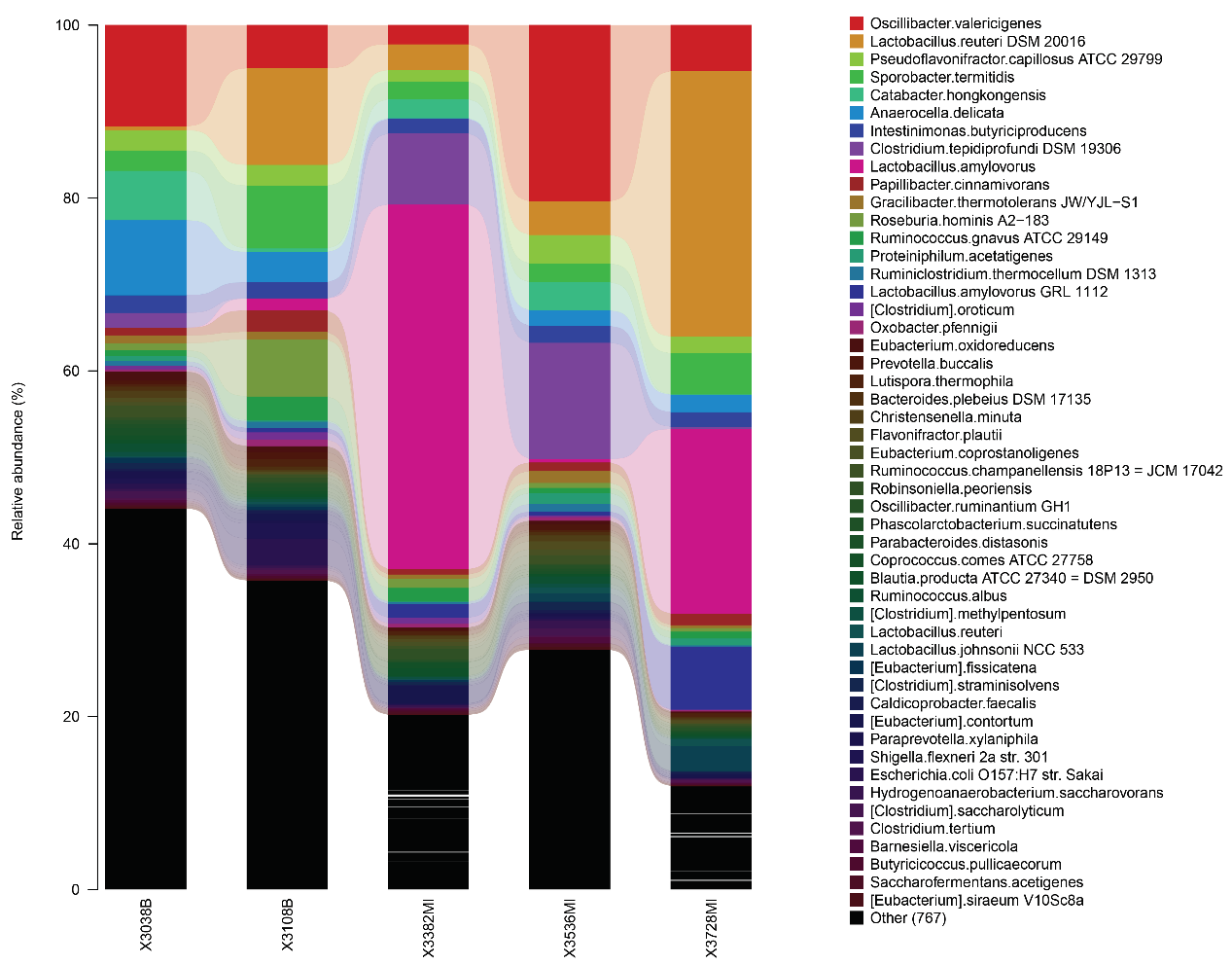
种水平的精细组成图
上图中,横坐标依据样本名排列,每一个柱形图代表一个样本,并以颜色区分各分类单元,纵坐标代表各分类单元的相对丰度,柱子越长,该分类单元在对应样本中的相对丰度越高。
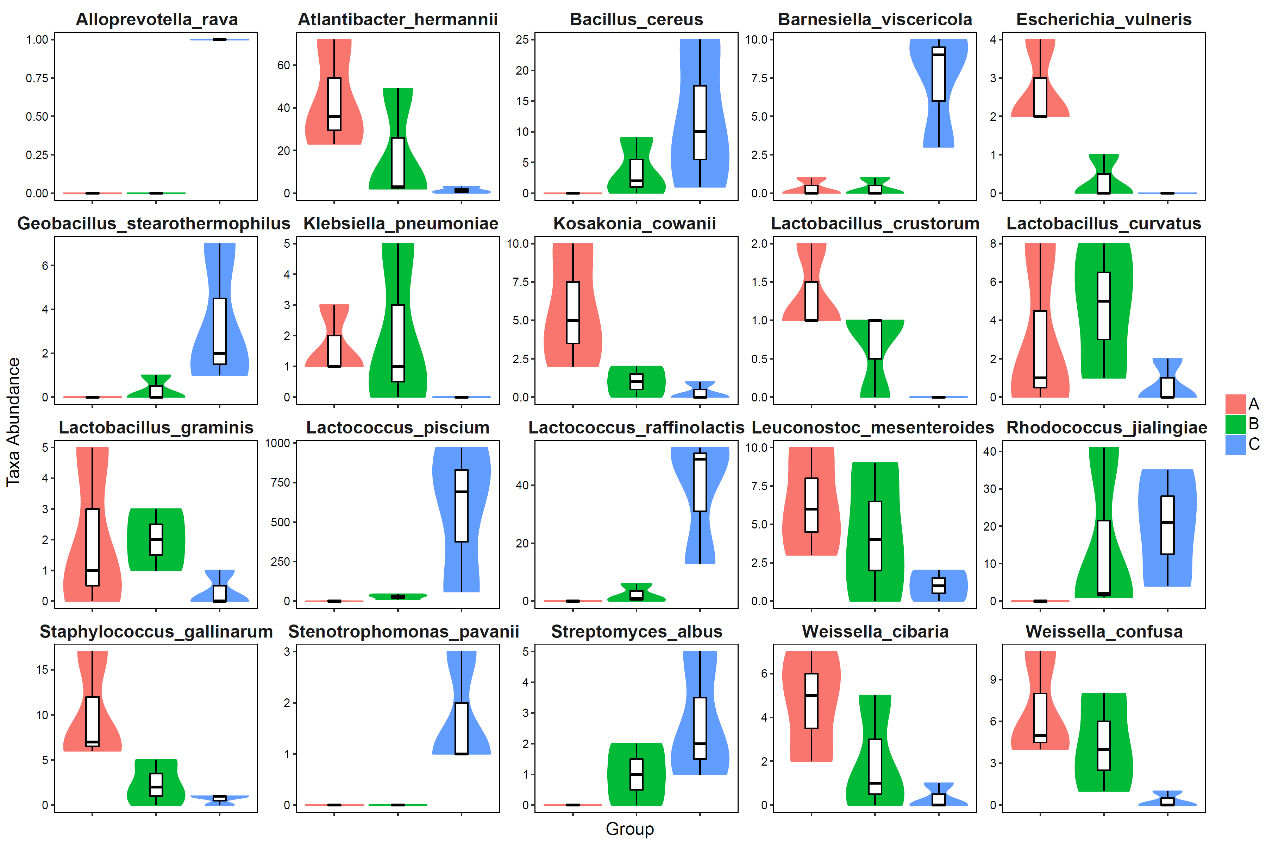
组间差异显著的种水平分类单元的丰度分布小提琴图
上图中,横坐标为组间差异显著的种水平分类单元,纵坐标为各分类单元在各样本(组)内的序列量,以小提琴图结合箱线图的形式展示:其中,小提琴图可以直观地显示数据的分布特征,“小提琴”的“胖瘦”反映了样本数据分布的密度高低(宽度越宽,表明该序列量水平所对应的样本越多);箱线图边框代表上下四分位数间距(Interquartile range,IQR),横线代表中位值,上下触须分别代表上下四分位以外的1.5倍IQR范围,符号“•”表示超过范围的极端值。
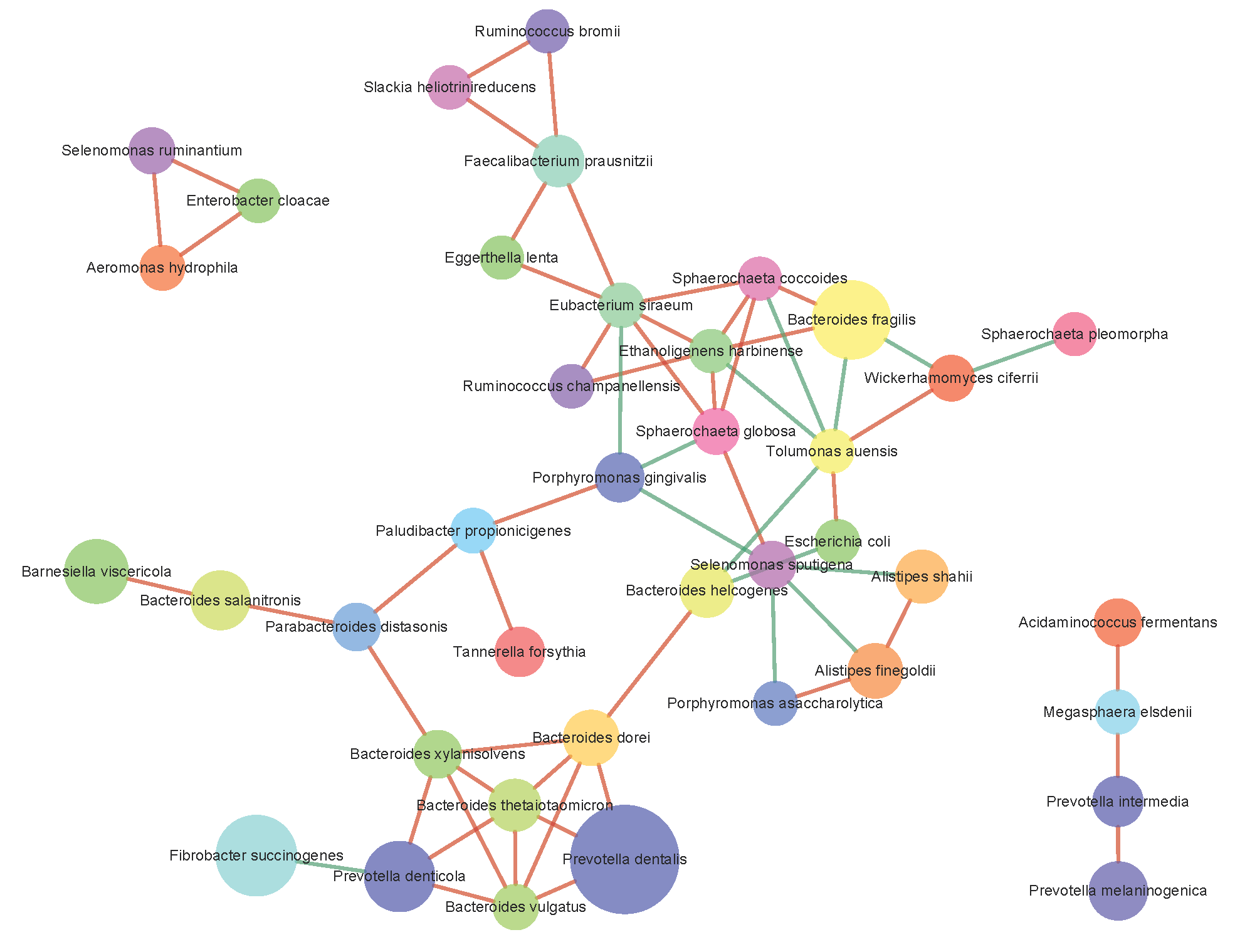
种水平关联网络分析图
上图中,各节点的圆圈代表种水平的各分类单元,以不同的颜色标识,节点之间的连接表明两个物种之间存在相关性,红线表明正相关,绿线表明负相关。通过某节点的连接越多,表明该物种与菌群中其它成员的关联越密切。
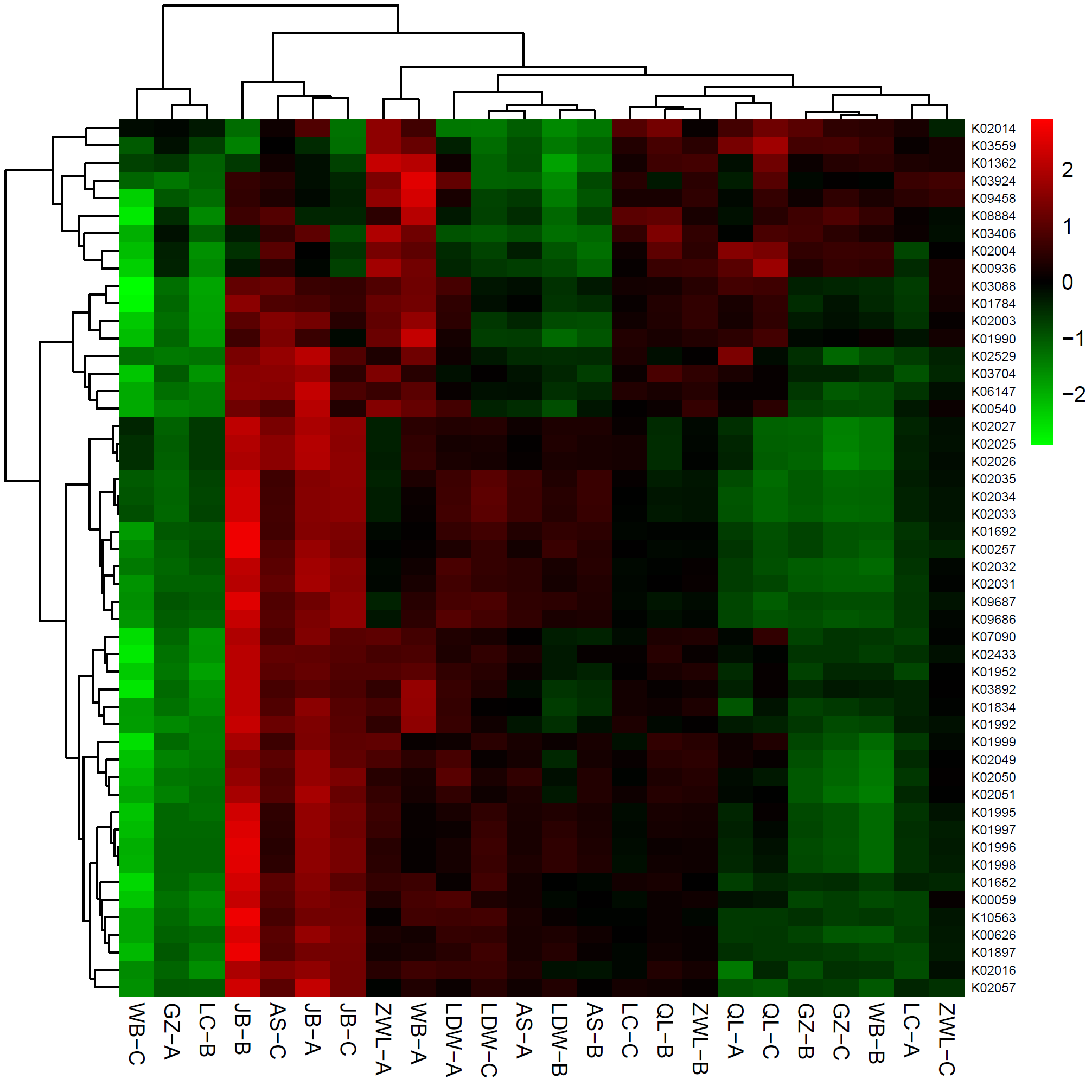
菌群代谢功能预测的KEGG直系同源基因簇(KO)丰度热图
上图中,样本先按照彼此之间功能类群丰度分布的相似度进行聚类,根据聚类结果横向依次排列。同理,功能类群也按照彼此在不同样本中分布的相似度进行聚类,根据聚类结果纵向依次排列。图中,红色代表在对应样本中丰度较高的功能类群,绿色代表丰度较低的功能类群。
利用三代测序技术对微生物组实现“高分辨率”解析

该研究于2016年发表于微生态领域顶级期刊《The ISME Journal》上(最新影响因子:9.328)。
研究背景
过去十多年中,随着短读长、高通量二代测序的兴起,以16S rRNA基因部分可变区序列为目标的分析方法,逐渐替代了基于rRNA基因全长克隆文库的一代Sanger测序法,使我们能对菌群组成进行深度定量解析。然而,由于二代测序读长短的特性,我们无法对测得物种进行精确的分类鉴定,从而限制了我们对菌群代谢功能的深入理解。随着三代SMRT测序技术的出现,我们有望从根本上解决这一瓶颈,实现对rRNA基因全长序列的高通量精准测序。
研究方法
样本来源:23种细菌和3种古菌组成的人工菌群,以及取自湖水的天然微生物群落样本。
测序平台:PacBio RS II(16S rRNA基因全长)+Illumina MiSeq(16S rRNA基因V4区)。
对两种测序平台的测序结果进行多样性组成谱分析,并通过计算机模拟,比较16S rRNA基因全长序列和单V区序列的物种分辨能力。
研究结果
根据两种平台的测序结果,门水平的菌群组成较为相似,但在更精细的水平存在差异。同时,三代测序的结果显著降低了物种分类信息的不确定性。计算机模拟的结果也表明,短读长的单V区测序可能严重低估某些特定类群的微生物,比如水体样本中参与氮循环和甲烷代谢的物种。因此,基于三代测序的菌群多样性组成谱分析能极大地提升物种分类鉴定的精确性,实现“高分辨率”检测的同时,也为深入阐释菌群的代谢功能奠定了基础。
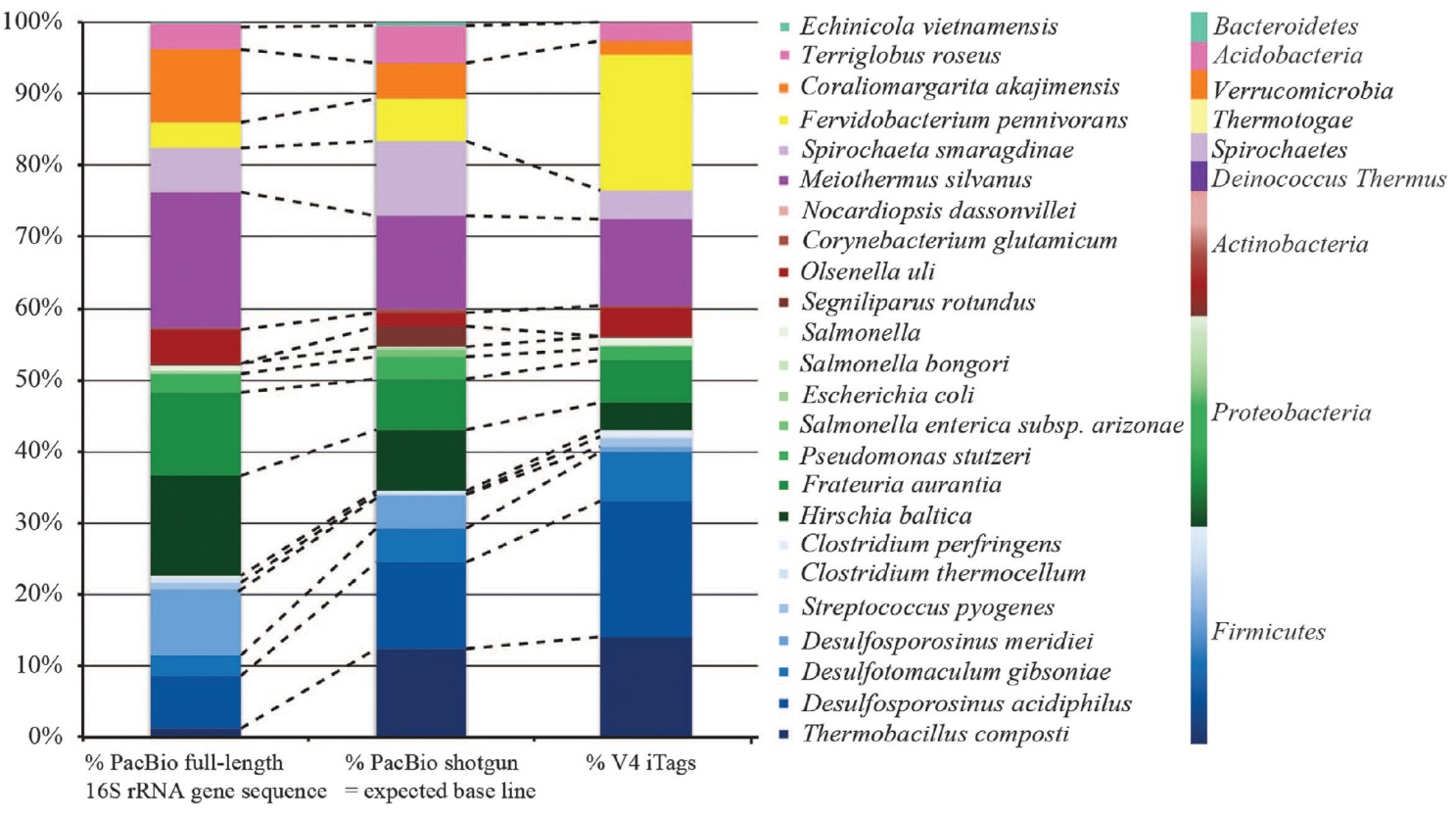
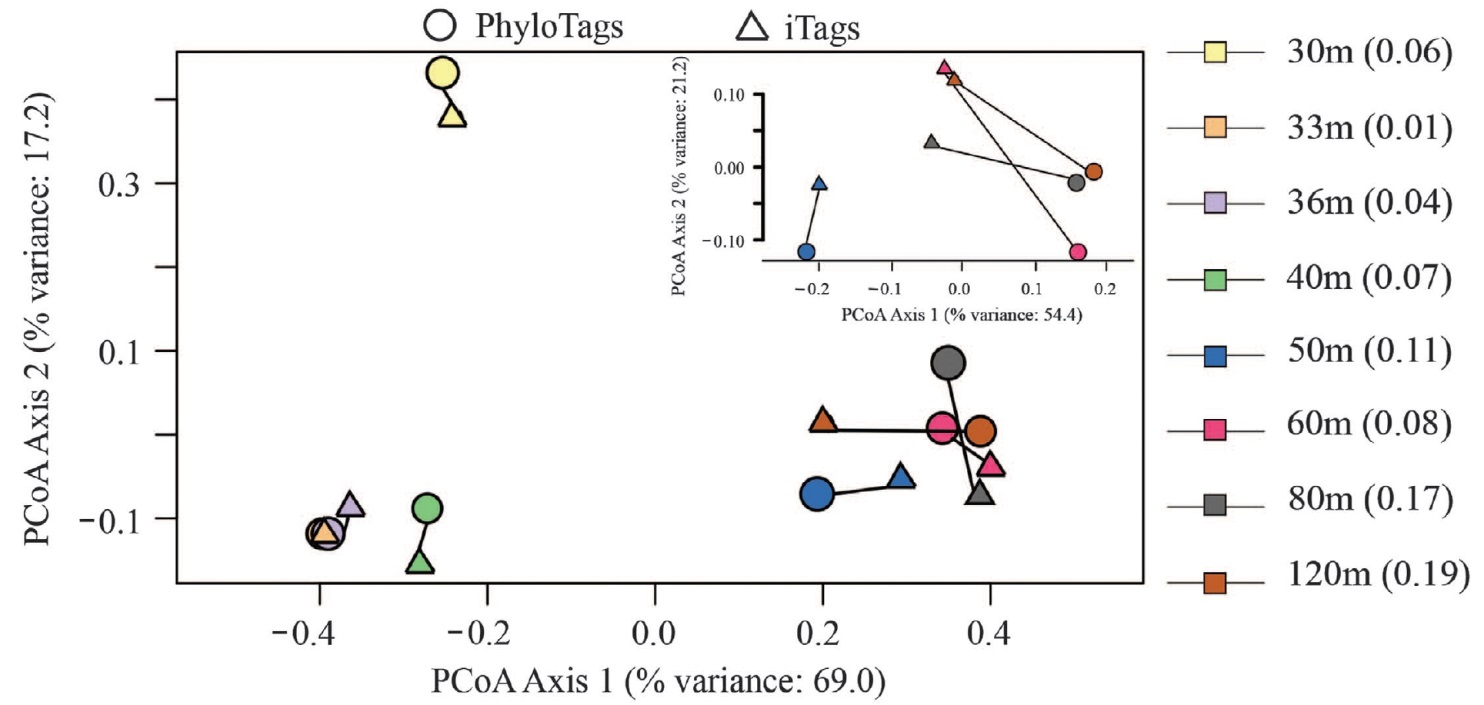
两种测序平台测序结果的整体比较。两者菌群的整体结构较为相似,但当菌群复杂程度增加时(底图子图),两者的检测结果有所差异。PhyloTags,三代PacBio RS II测序结果;iTags,Illumina MiSeq测序结果。
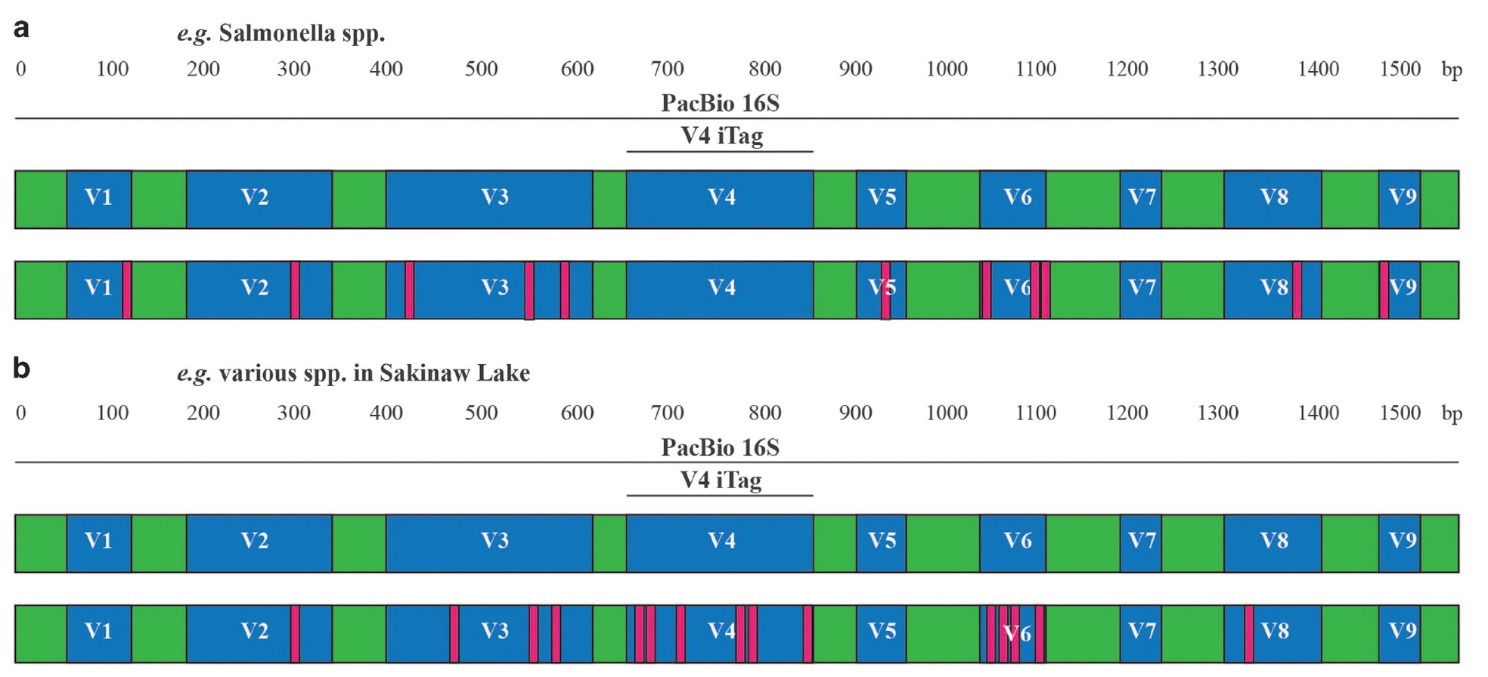
16S rRNA基因各V区的序列保守性并不一致,因而基于16S rRNA基因全长序列的三代测序,可以更全面地反映物种的种属信息。a图,以Salmonella属为代表,全长序列的种间差异达到97.4%,但V4区高度保守,二代测序结果将低估该物种的多样性;b图,某些物种在V4区的多样性可能高于其他V区,因而二代测序结果将高估相关物种的多样性。
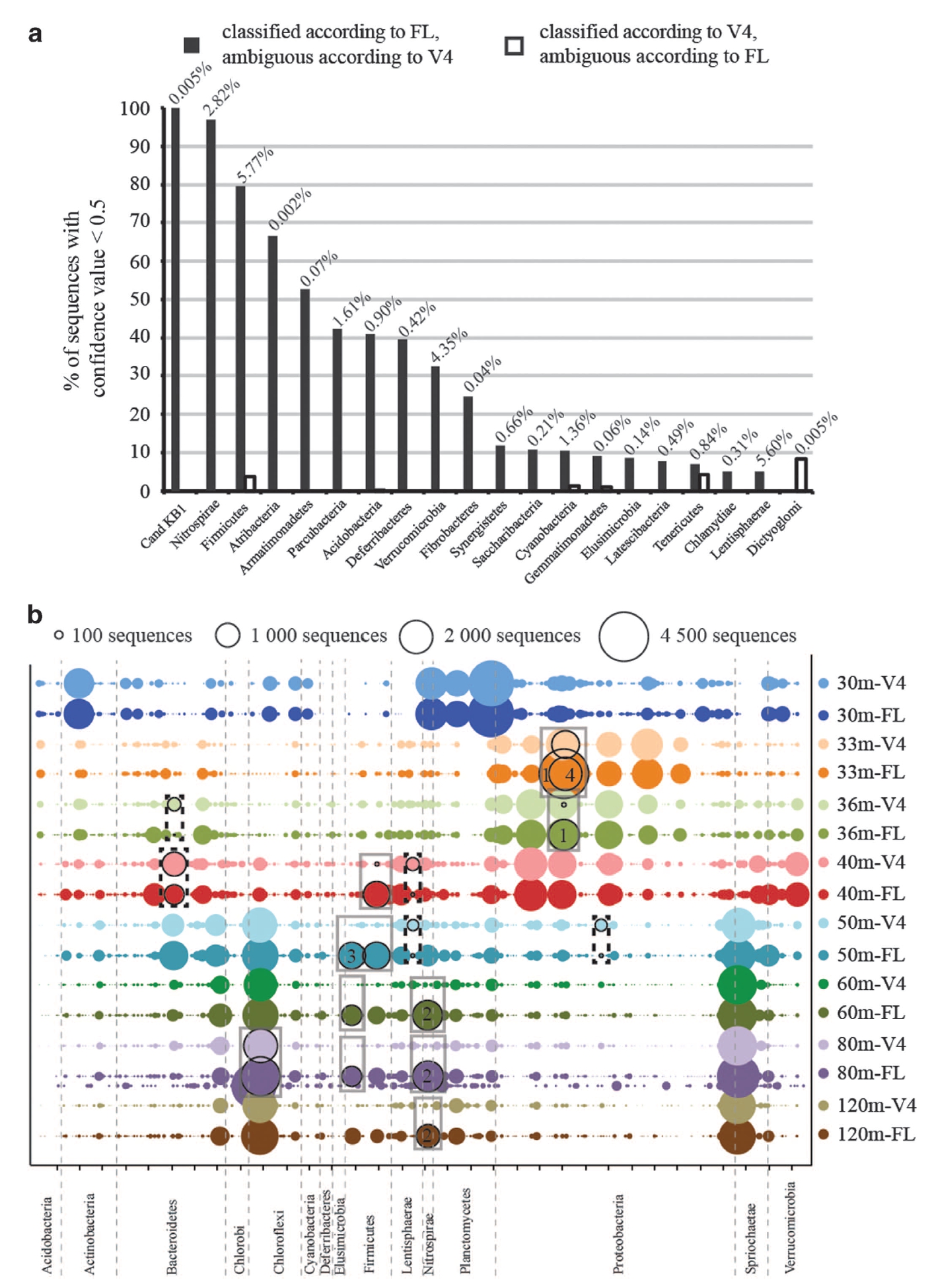
两种测序平台测序结果的精细比较。a图,三代测序的结果显著降低了物种分类信息的不确定性;b图,二代单V区测序结果可能会严重低估某些特定微生物类群的含量(以方框标记)。
研究结论
综上所述,运用三代测序技术进行微生物组研究,将显著提升菌群多样性组成谱解析的精准性和全面性。
参考文献
Singer, E., Bushnell, B., Coleman-Derr, D., Bowman, B., Bowers, R.M., Levy, A., Gies, E.A., Cheng, J.-F., Copeland, A., Klenk, H.-P., et al. (2016). High-resolution phylogenetic microbial community profiling. ISME J 10, 2020-2032.
虫洞平台上的所有商品信息、客户评价、商品咨询、网友讨论等内容,是虫洞时空重要的经营资源,未经许可,禁止非法转载使用。
注:本站商品信息均来自于合作方,其真实性、准确性和合法性由信息拥有者(合作方)负责。本站不提供任何保证,并不承担任何法律责任。
批发价/零售价/专属价:均为商品的销售价,不同性质的单位所看到的价格会有所不同,是您最终决定是否购买商品的依据。 划线价:商品展示的划横线价格为参考价,并非原价,该价格可能是品牌标价或由品牌供应商提供的零售价(如厂商指导价、建议零售价等) 或该商品在虫洞平台上曾经展示过的销售价;由于地区、时间的差异性和市场行情波动,品牌专柜标价、商品吊牌价等可能会与您购物时展示的不一致,该价格仅供您参考。
异常问题:商品的具体售价以订单结算页价格为准;如您发现商品售价或促销信息有异常,建议购买前先联系卖家咨询。
北京虫洞时空于2021年10月成立,为进一步提升实验室供应链效率, 解决实验室在采购中存在的选货、价格、物流和结算难等问题,提升实验室服务的互联网化水平,公司致力于成为中国领先的专业实验室第三方电商平台。











